
Docs
Here we provide a brief description of the analyzed parameters and used definitions. For details and references please see the MINT manual.pdf.
Hydrogen bonds and base pairs
A hydrogen bond is formed if a hydrogen atom is placed close to an acceptor atom, and if the angle between the donor atom, hydrogen and acceptor is ideally close to 180 degrees (Figure 1). The user may define both parameters: minimal angle and maximal distance between the acceptor and donor or the acceptor and hydrogen.
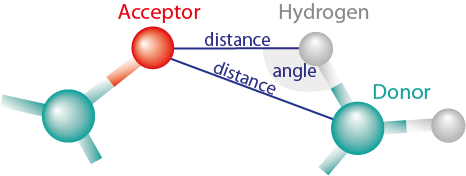
Figure 1: Hydrogen bond is detected when the distance is smaller then cutoff and angle (created by Acceptor, Hydrogen and Donor) is larger than cutoff angle. MINT can measure distance between both Acceptor and Donor, and Acceptor and Hydrogen.
We defined a list of all possible acceptors and donors for all four nucleotides. The acceptors and donors are assigned to the edges of the nucleotide, as shown in Figure 2. MINT determines the interacting edges of every pair of nucleotides from the names of the interacting atoms. However, several atoms are placed in the corners of a molecule and therefore participate in more than one edge. In such cases, the rest of bonds are classified, and the prevailing edge is chosen. If there is only one hydrogen bond, both names are returned.
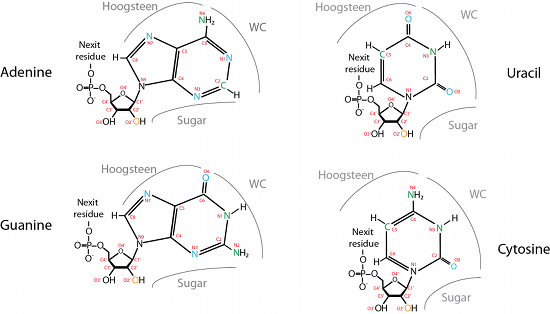
Figure 2: Every nucleotide has three sides/edges that can create hydrogen bonds with another nucleotide[1].
After detecting the hydrogen bonds and defining the interacting sites, the cis/trans configuration is computed. MINT measures the torsion angle formed by the four atoms and depending on its value, the configuration is assigned. Both cis and trans configurations are shown in Figure 3.
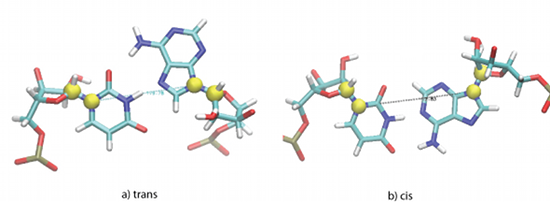
Figure 3: Yellow spheres correspond to atoms: C1' and N1 in pyrimidines or N9 in purines. The torsion angle created by these four points determines the configuration of the two nucleotides creating a pair.
MINT maps all hydrogen bonds for all nucleotide pairs that are within the cutoff distance. The cutoff is measured between the C1' carbons of every nucleotide.
In the output description file the user will find description of the WC-WC bonds with atom names and distances
WC Base pairs:
0] U2/A116 N3-H3-N1 angle: 150.65 distance: 1.91 | N3-H3-C2 angle: 164.89 distance: 2.48 | O4-H61-N6 angle: 151.98 distance: 1.82 WC/WC/3 Cis
1] C3/G115 N4-H42-O6 angle: 147.3 distance: 2.26 | O2-H22-N2 angle: 168.89 distance: 2.01 | N3-H1-N1 angle: 166.09 distance: 2.08 WC/WC/3 Cis
2] U4/A114 N3-H3-N1 angle: 169.96 distance: 1.83 | N3-H3-C2 angle: 156.01 distance: 2.65 | O4-H61-N6 angle: 151.51 distance: 2.23 WC/WC/3 Cis
3] U5/A113 N3-H3-N1 angle: 167.78 distance: 1.91 | N3-H3-C2 angle: 160.28 distance: 2.5 | O4-H61-N6 angle: 150.16 distance: 2.42 | O2-H2-C2 angle: 140.25 distance: 2.76 WC/WC/4 Cis
and non-WC-WC pairs:
Other interactions:
0] G0/C118 N2-H22-N3 angle: 168.54 distance: 1.89 WC*Sugar/WC/1 Cis
1] U1/A117 O4-H61-N6 angle: 162.14 distance: 1.71 WC*Hoogsteen/WC*Hoogsteen/1 Cis
2] A12/C80 C2-H2-O2 angle: 167.64 distance: 2.17 WC*Sugar/WC*Sugar/1 Trans
Motifs
MINT detects all kinds of motifs that are defined as a set of unpaired nucleotides with the surrounding pairs.
For clarity MINT uses the numerical description of the motifs. Figure 4(b) shows a four nucleotide loop at the end of a hairpin, represented by a single number 4. Figure 4(c) shows a symmetrical bulge loop, with bulge length of three and is represented by two numbers: 0-3. The three-way junction, shown in Figure 4(d), without unpaired nucleotides is represented by the three zeros: 0-0-0. The numbers correspond to the number of unpaired nucleotides between pairs. Therefore the multibranched loop in Figure 4(e) is denoted as 1-2-3.
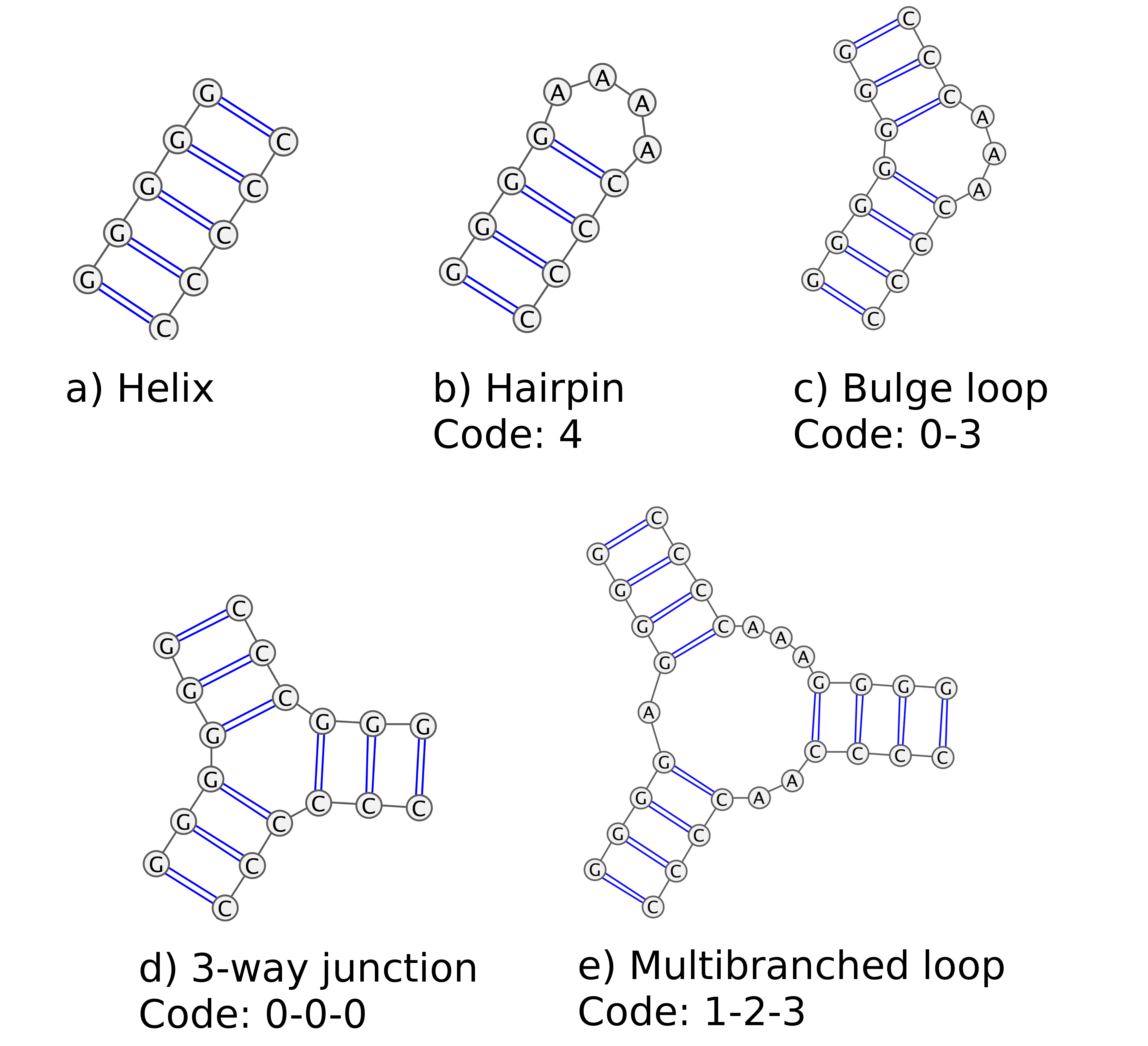
Figure 4: A scheme representing several types of structures occurring in RNA with their MINT codes. Pictures generated using Varna applet.
MINT describes all secondary structural motifs and lists them in the _description output file. First, the output lists all the helices:
Helices:
1] G500-C504 -> G541-C545
2] C511-G515 -> C536-G540
3] A520 -> A533
4] G521-C522 -> G527-C528
then MINT provides a list of motifs than can be easily visualized in the VMD program in its graphical representation window:
Helices vmd
1] resid 500 to 504 541 to 545
2] resid 511 to 515 536 to 540
3] resid 520 520
4] resid 521 to 522 527 to 528
The same is done for all other motifs:
Motifs:
1] 3-0-16 C8-G110-A109-A108-G107-G106-C90-G89-C25-A24-G23-G22-G21-A20-G19-A18-C17-G16-A15-A14-G13-A12-G11-A10-A9-C8-
2] 4-14 G28-C86-U85-A84-A83-A82-U81-A43-C42-U41-U40-C39-G38-A37-A36-G35-C34-A33-G32-C31-C30-C29-G28-
3] 1-2 C45-G79-U78-G77-C48-A47-A46-C45-
...
Pseudo-knots
From all Watson-Crick pairs, a list representation of the RNA secondary structure is created. Each nucleotide may have only one Watson-Crick partner. If the second WC-WC partner is encountered all three nucleotides are added to the triplets list. Index of the list represents the number of the nucleotide and the stored value is the number of its Watson-Crick partner.
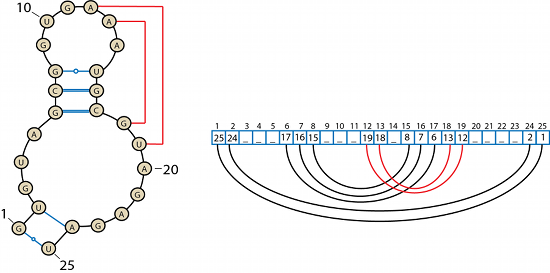
Figure 5: An example of a secondary structure of an RNA molecule and its list representation. Red lines correspond to the Watson-Crick interactions creating a pseudo-knot.
The internal representation of the RNA structure contains also information about other higher-order structures such as pseudo-knots. A pseudo-knot is also formed by the Watson-Crick interactions but creates three-dimensional folds. MINT detects the pseudo-knot fold when the arcs intersect. A pseudo-knot is a symmetrical structure - three pairs 6-17 7-16 8-15 shown in Figure 5 form the pseudo-knot as well as two pairs: 12-19 13-18; the natural way of solving this conflict is to choose the shorter list, in that case pairs 12-19 13-18.
In the _description output pseudo-knots will be described as following:
Pseudo-knots:
G13-C42 A14-U41 A15-U40 G16-C39 C17-G38 G21-C31 G22-C30 G23-C29 U85-A109
G13-C42 A14-U41 A15-U40 G16-C39 C17-G38 G21-C31 G22-C30 G23-C29 U85-A109
Triplets
If there is more than one partner for a certain nucleotide, MINT puts them in the Triplets list. Exemplary triplet is shown in Figure 6.
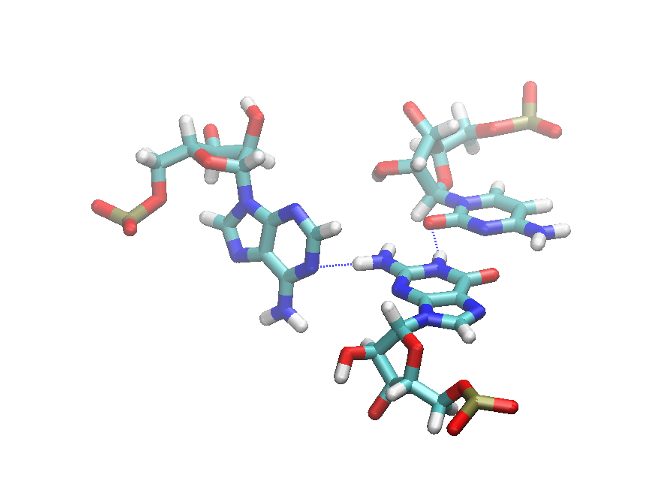
Figure 6: The exemplary triplet shown in VMD with hydrogen bonds marked as blue dashed lines.
Triplets will be enlisted in the _description file as follows:
Triplets:
1] A1014-G987-C1218-
Triplets vmd
1] resid 1014 987 1218
Stacking
We estimate stacking between two bases by calculating the energy of electrostatic (Coulomb) and van der Waals interactions, applying the equations used in molecular mechanics. We consider two bases as stacked if the value of the VDW interaction energy between them is smaller than a given parameter vdw_cutoff_stacking by default set to [-0.5 kcal/mol]. Only pairs that satisfy this condition will be printed in the _description and _stacking_in_time.csv files.
Calculations of the interaction energy are performed only for pairs for which the distance between the centers of mass of the bases is smaller than user defined cutoff stacking (default 5.5 [Å]). An example of the stacking pairs is shown in Figure 7.
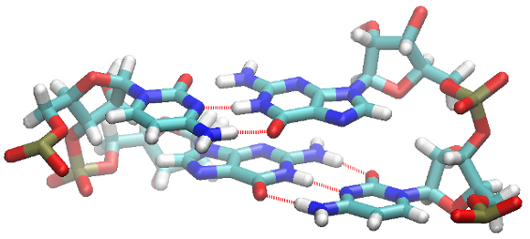
Figure 7: Two WC-WC basepairs in a stacked configuration.
In the description_ output the stacking pairs are outlined as follows:
Stacking pairs
n1 n2 Coul VdW sum
G0 U1 (2.62, -6.0, -3.38)
G0 U2 (1.06, -0.3, 0.76)
U1 U2 (3.86, -3.99, -0.13)
MINT can also detect the interactions termed ion-pi. This is an arrangement in which one of the phosphorus atoms is placed close to a nucleotide ring, as presented in Figure 8. For details and references please see the MINT manual.pdf.
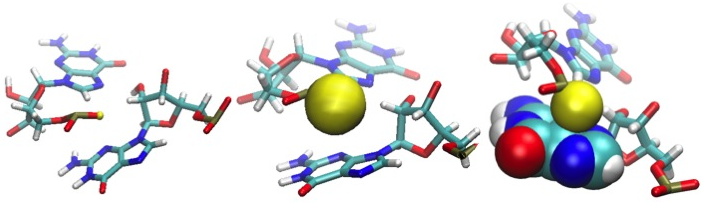
Figure 8: Three representations of the oxygen atom (yellow) "stacked" over the guanine base. The yellow sphere corresponds to the real van der Waals radius.
The list of these conformational arrangements will appear in the _descriptiom output file:
Stacking Pi
base1 base2 OPatom_from_base2 avg_distance coulomb_energy vdw_energy energy_sum
C1336_A1238_O1P [4.37, (-19.49, 44.38, 24.89)]
G1077_G1079_O2P [3.81, (10.57, 0.5, 11.07)]
G1266_G1268_O2P [3.63, (11.54, 0.43, 11.97)]
References
[1] Lescoute and E Westhof. The interaction networks of structured RNAs. Nucleic acids research, 34(22):6587-604, January 2006.