
FAQ
How can I cite MINT?
If you find MINT useful in your research, please contribute to its manuscript:
Górska A, Jasiński M, Trylska J. MINT: software to identify motifs and short-range interactions in trajectories of nucleic acids. Nucleic Acids Res. 2015 May 29. doi: 10.1093/nar/gkv559 PubMed PMID: 26024667.
Górska A, Jasiński M, Trylska J. MINT: software to identify motifs and short-range interactions in trajectories of nucleic acids. Nucleic Acids Res. 2015 May 29. doi: 10.1093/nar/gkv559 PubMed PMID: 26024667.
How are modified nucleotides represented in MINT?
For modified nucleotides MINT uses a set of Amber force field parameters from J. Chem. Theory Comput., 2007, 3 (4), pp 1464–1475 . For other modified nucleotides, for which MINT has no parameters, the stacking energy values are set to 0 and all recognized hydrogen bonds are classified as belonging to the non-Watson-Crick base pairs.
If you have an RNA molecule with nonstandard nucleotides that you have parameterized on your own, you should download MINT and use it locally. You have to modify both the nucleotides.csv and charges_andVDW_DNA.csv file (in the MINT/data/ directory) introducing your own parameters.
Why sometimes the orientation of glycosidic bonds is cis instead of trans ?
Sometimes the orientation of the glycosidic bonds (cis or trans) obtained by MINT differs from what was given for a particular base pair conformation in:
Nucl. Acids Res. (2002) 30 (16): 3497-3531 .
One has to consider that this classification is based on fixed X-ray structures of RNA molecules.
MINT cis/trans recognition relies on the measurement of the torsion angle formed by four atoms (C1’, N1 in pyrimidines and C1’, N9 in purines).
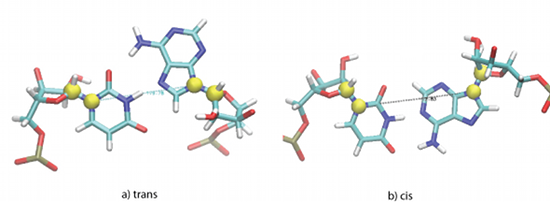
Figure 1: Yellow spheres correspond to the C1' atoms and N1 in pyrimidines or N9 in purines. The torsion angle created by these four points determines the configuration of the two nucleotides creating a pair.
In molecular dynamics (MD) simulations molecular internal motion is propagated so it is possible that for specific base pairs even a small change in the arrangement of one of the bases changes the overall cis/trans orientation (e.g. A-G Trans Hoogsteen - Sugar Edge).
Why sometimes MINT finds in canonical base pairs more hydrogen bonds than expected?
The analysis of hydrogen bonds in MINT is based only on geometric criteria (distance and angle). To detect a hydrogen bond the default maximal distance between a donor and acceptor is set to 3.5 Å and the minimal angle created by the donor, hydrogen and acceptor is 150 degrees. It is possible that in the crystal structures or user-provided structures more than three hydrogen bonds in a GC pair (see Figure 2) or two in AU pair will satisfy the above criteria. The user can remove the "unwanted" hydrogen bonds by decreasing the distance and/or increasing the angle. However, one should have in mind that too strict criteria can cause errors in the analysis of nucleotide pairs and assignment of the RNA secondary structure.
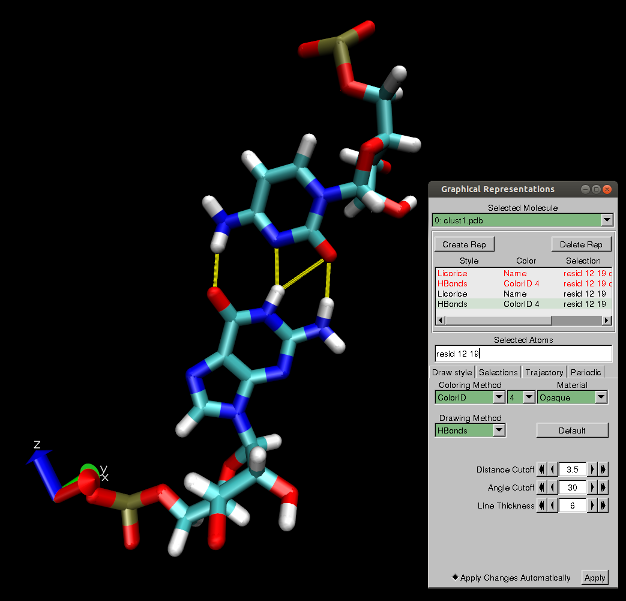
Figure 2: Visualization of four possible hydrogen bonds (yellow) found in the GC pair.
Figure 2: Visualization of four possible hydrogen bonds (yellow) found in the GC pair.